Final Report/Thesis 2019
Abstract
Somerton Man case is most mysterious case in last century. A unknown man was murdered on Somerton Beach, and identifications of the killer and the victim are still mysteries nowaday. The project aims to investigate the identification of the Somerton man with his DNA data provided. Unfortunately, the DNA data is corrupted and has a high drop rate, therefore the team of the project would be required to use different strategies and techniques to recover and analyse the DNA. Then find out any possible characteristics of the Somerton Man.
To approach the goals of the project, the team would have firstly evaluate the DNA data and try to conduct DNA analysis via different genetic services. In addition, the degradation process of complete DNA data. By degrading a complete DNA file, how the degradation would affect the DNA analysis results would be observed and discussed.
Introduction
Motivation
The main topic of the project is human identification via using software programming and genetic analysis techniques. The project conducts a study on investigating the identification of the victim in the Somerton Man case which is one of the most mysterious cases in last century. On December 1st 1948, a well-dressed male was found dead on Somerton Beach in Adelaide [1]. He was clean-shaven, well dressed in a suit and no belongings could prove his identity [7]. Later the man was called as Somerton Man. The figure below shows the look of the Somerton Man.

After more than half century, the identification of the Somerton Man is still unsolved. With the Somerton Man's DNA data extracted from his hair, the project team may be able to conduct several DNA examinations and identify characteristics of the man. Unfortunately, the DNA data is corrupted due to degradation of the hair, but the team of the project will try the best to investigate the data with modern techniques. In addition, human identification is the main topic of the project. In modern society, human identification techniques is useful in multiple aspects, such as criminal investigation or seeking relatives. Most current identification techniques would require high quality DNA samples, but the project focus on investigating identification techniques based on low quality genetic data. Also the project concentrate on using engineering methods and problem solving skills to improve the identification techniques.
Objectives
The aim of the project is to investigate the identification of the Somerton Man. To be more specific, the group is aiming to identify any possible relatives, physical characteristics, genetic diseases or ethnicity of the Somerton Man. To achieve these goals, the team would use software and genetic analysis techniques to work on the Somerton Man's DNA data. In addition, the reliability of DNA analysis results from a low quality DNA data would be investigated. This would be approached by degrading several complete DNA data samples into different levels and conducts sets of genetic test on them. The change of test results will be observed and discussed.
Background
DNA
DNA is the hereditary material which stores the genetic information in humans [2]. There are two types of DNA in human beings, one is known as nuclear DNA which is located in cell nucleus and another type is mitochondrial DNA which is located in the mitochondria. This project only focuses on the analysis of nuclear DNA. DNA stores genetic information as a sequence built up with four types of nitrogen bases which are adenine (A), guanine (G), cytosine (C), and thymine (T) [2]. Also, a sugar molecule and a phosphate molecule are attached to each nitrogen base to form a molecule called nucleotide. The bases would pair up (A with T and C with G) and multiple nucleotides are placed in two strands to form a double helix which looks like a spiral [2]. In general, a DNA is a genetic sequence formed by multiple base pairs. The genetic instructions of building and maintaining an organism are obtained from the order of these base pairs [2]. There are about 3 billion bases in human DNA, in which more than 99% of the bases are common in all human beings, and the physiological differences among people depends on these 1% DNA.

Chromosome


Chromosome is an integrated package of DNA molecules. It has thread-like structure, and DNA molecules are coiled up around hi stones proteins to form the structure [3]. There are 23 pairs of chromosomes in human body’s cell, which is 46 chromosomes in total. 22 pairs are called autosomes which are common for both males and females and the last 23rd pair is sex chromosomes which differ males and females. In this project, the DNA data analysis would only focus on autosomes [4].
SNP
Single nucleotide polymorphisms(SNPs) are most common type of genetic variation among human beings [5]. Each SNP represents a difference in a nucleotide which is a single DNA molecule [6]. For instance, a SNP may replace a nucleotide of base guanine (G) with cytosine (C). These SNPs can be found nearly once in every 1,000 nuceotides on average in a person’s DNA. Most SNPs do not effect health of owner. However, some of these variations may associated with diseases.
DNA reference file
A DNA reference file stores a group of SNPs data of owner’s DNA. The format of DNA reference files using in this project is the same format of 23andMe company’s file, where 23andMe is a company that attended to provide personal genetic information for the customer by using advanced genetic analysis techniques and web-based interactive tools. A screen shot of a sample file is shown below.

As shown in the figure, there are 4 columns rsid, chromosone, position and genotype in the DNA reference file. The rsid is a unique id used to identify a specific SNP [9]. The format of rsid starts with “rs” and followed by a number (eg. rs123456). These rsids are commonly used by researchers and databases. There is another special format of rsid that starts with “i” and followed by a number (eg. i123456). This “i” format is used internally by 23andMe to identify the unknown SNP and can not be used in public database. The second column chromosone identify which chromosome the SNP belongs to. Then the third column position indicates positions of SNPs in owner’s DNA sequence. Last column genotype represent the base pairs of variants(A, T, G, or C). Note that there are some cases, the genotype result for some SNPs are not able be provided and “--” would be displays in genotype column [9]. It is important to note that only the SNPs with identified base pairs can be used for DNA analysis.
Task 1
Aims
The aim of this task is to have a basic understanding of the DNA reference file and DNA analysis techniques. The project provide a DNA reference file of the Somerton Man which is a corrupted DNA data. A screen shot of the file is shown in figure 6.

The first goal of this task is to evaluate the quality of the file including counting the total amount of SNPs and the amount of available SNPs. Then the team should try to conduct some DNA analysis on the DNA reference file.
Methods
To approach the first goal of this task, the team will develop a program which provide functions for counting total amount of SNPs, amount of available SNPs (SNPs that do not have genotype of “--”) and determine the percentages of available SNPs for 1 to 22 chromosones of Somerton Man’s DNA raw data. Program was developed by C++ language. Then a website called GEDmatch will be used for conducting DNA analysis. GEDmatch is a website that has an open data personal genomics database and provide tools for DNA and genealogy research. The site become well known after law enforcement in California use it to the Golden State Killer case and are commonly used by all law enforcement in United State [10]. Somerton Man’s DNA reference file will be uploaded to the website and tried to conduct several DNA analysis provided on the website.
Results and discussion
The counting outputs of Somerton man's DNA data is presented in figure 7. As the figure shown, there are more than 0.6 million SNPs in the files, but only about 2% of them have determined base pairs. In DNA analysis, only the SNPs with available base pairs can be used and most genetic analysis technique would require a certain amount of available SNPs.

Then the Somerton Man's DNA reference file was uploaded to GEDmatch for using one-to-many tool. The one-to-many tool is the main service provided by GEDmatch. When a DNA raw data reference file was updated, it would be stored as a kit in GEDmatch database, and be compared with other kits in the public database. After the match process finished, the one-to-many tool could show how many kits in database are matched with the kit that the user has uploaded. Unfortunately the website reject to process the Somerton Man's data for using one-to-many tool since the file did not meet the minimum requirements of 2000 SNPs for each chromosome.

Conclusion
The quality of Somerton Man's DNA reference file is lower than expected. Only about 2% of 613905 SNPs in the files are available for use. Such low quality DNA file is not accepted by GEDmatch to conduct DNA match examination. In order to satisfy the minimum requirements of GEDmatch, a data recovery work would be required which would be introduced in next task.
Task 2: Artificially recover DNA file
Aims
In this task, the project group aims to artificially recover Somerton Man’s DNA file to satisfy the basic SNPs amount requirements (2000 SNPs for each chromosome) of GEDmatch’s one-to-many tool and find out how many people is relative with Somerton Man’s DNA kit.
Methods
The recovery works would be done by developing multiple programs with C++. In general, the recovery work is to replace fixed amount of empty SNPs which is 2000 SNPs for each chromosome with available SNPs. Several simple recovery algorithms will be introduced and implemented. First algorithm called random algorithm is to replace empty SNPs with random base pairs in genotype. Replacing empty genotype with homozygous pairs (AA, GG, TT, CC) can be considered which provide 4 new algorithms to use. In addition, if there is no DNA kit matched with Somerton Man’ DNA in the database, trying to recover Somerton Man’s DNA more empty SNPs can be a back up plan. With the recovery algorithm introduced before, the project team can recover more SNPs in Somerton Man’s DNA reference file. And try the recovered DNA kits with one-to-many tool.
Results and discussion
With the developed program, multiple artificial DNA kits which have 2000 SNPs in each chromosome were created. Unfortunately, all of these DNA kits have 0 matches with other DNA in the public database which means these artificial DNA kits have no relative can be found in the GEDmatch database.

Then kits with more amount of empty SNPs were replaced with homozygous pairs or random pairs were created, but none of these files could find relative DNA kits in the database. Even the DNA kits with all empty SNPs recovered could find a matched DNA result. It is important to note that all 5 recovery strategies were all implemented. As GEDmatch is the most commonly used DNA database in public, it contained a huge amount of DNA kits in its database. As the website shown, the total number of kits managed by GEDmatch database is 1363427157376. Therefore, the chance that no DNA kit in the database is related to Somerton Man is nearly impossible. Which means that the quality of Somerton Man’s DNA file is too low for using one-to-many tool and implementing simple recovery algorithms introduced are useless.
Conclusion
It is obvious that there is too many empty SNPs in Somerton Man’s DNA reference file. And the recovery algorithms introduced in section 4.1 are too simple and can not help to increase the chance of finding Somerton Man’s relatives in the GEDmatch database. With DNA data that only contains approximate 2% available SNPs, it is nearly impossible to find any possible related DNA kits with Somerton Man.
Task 3: Investigation on ethnicity
Aims
The first aim of this task is investigating the ethnicity of Somerton Man’s DNA. As described in previous section, the quality of Somerton Man’s DNA is low, therefore the second aim is to study the reliability of low quality DNA’s ethnicity examination results.
Methods
Firstly, an ethnicity tool called Eurogenes Ad-Mix Utilities will be used. This tool is provided on GEDmatch and can generate a report of ethnicity proportions with given DNA kit. Eurogenes K13 model is selected as the 'calculator' model. This model allow the utility calculate the ethnicity proportion into 13 different global regions as figure 10 shown, and this mode is primary for European background persons since it provide more sub-continental regions for Europe [13]. The Somerton Man’s DNA would be selected as input kit of the utility and the ethnicity report will be generated and evaluated.

In addition, to investigate the reliability of a low quality DNA data file's ethnicity report, several complete DNA samples would be required. The project will order 2 sets of complete DNA reference data from 23andMe which provide same format as Somerton Man’s file. A program will be developed that allow the user to degrade the selected DNA file to be degraded into different levels of DNA data. This program will be also developed in C++. The project team will degrade each complete DNA sample files into 9 files by removing 10% SNPs, 20% SNPs and then step by step to 90% SNPs. An extra file which would only contained the SNPs with same rsids in Somerton Man’s DNA file and be named as degraded_DNA will be generated for each set of complete DNA sample data as well. Then these files will be uploaded to GEDmatch and conduct the same ethnicity research as what has been done on Somerton man’s DNA raw data. All ethnicity reports are going to be recorded, and the change of how the ethnicity proportion changes will be observed. In order to provide stronger evidence to prove whether the low quality DNA file's ethnicity report is reliable or not, different degradation algorithms are introduced to be performed. The first strategy is that for every 10 SNPs, remove first n% SNPs where n% is the percentage of SNPs we like to remove. The next algorithm perform opposite way of the first algorithm. This algorithm require the program to remove last n% SNPs for every 10 SNPs, where n% is the percentage of SNPs we like to remove. The third and fourth methods are remove the first and last n% of SNPs for each chromosome, where n% is the percentage of SNPs we like to remove.
Results and discussion
The ethnicity report of Somerton Man's DNA are shown in figure 11. As the pie chart shown, the first 2 major regions are North Atlantic region which has 36.21% proportion and Baltic region which has 20.44% proportion.

According to the population averages table[15] for Eurogenes K13 model provided by the developer Davidski (Polako), both Baltic and North Atlantic regions are in Europe. the figure 12 is a map that indicate the areas of Baltic region and figure 13 shows the North Atlantic region.
-
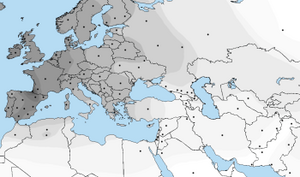
Figure 12: Map of Baltic region -
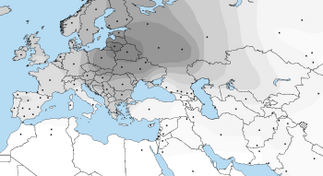
Figure 13: Map of North Atlantic region


To prove the ethnicity report created is reliable, 2 complete DNA files were gained and be degraded to the same level of Somerton Man’s DNA which is 2% SNPs remaining in the file. Sample DNA reference file 1 contained 613967 SNPs and 96.41% of them are not empty, and DNA reference file 2 has 614009 SNPs and 97.68% of them are available for use. The ethnicity reports of 2 complete sample DNA files are presented in figure 16 and 17. Also, ethnicity reports of degraded_DNA files for each complete DNA are shown in figure 14 and 15. According to the ethnicity reports shown in figures, the proportion of largest and second largest ethnicity regions of sample DNA file 1 have changed to 83.13% to 78.66% and 14.82% to 18.14% after degradation process. The first major region proportion has reduced 4.64% and the second region proportion has increased for 3.32%. The degradation process effect the proportion of each ethnicity region for DNA sample 1, but the change is not much and the first and second regions are still the largest 2 regions in the pie chart. Similar phenomenon can be discovered when comparing ethnicity reports of DNA sample 2. The largest ethnicity regions has grown for 2.33% from 81.44% to 83.77%, and the second largest region increased 0.28% from 7.12% to 7.40%. These changes shows that the proportion of major ethnicity regions would not have great change which could be within 5% when a complete human DNA file is degraded to a level of 2% SNPs remaining.
-
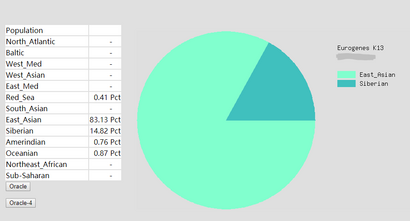
Figure 14: Ethnicity reports of sample DNA file 1 -
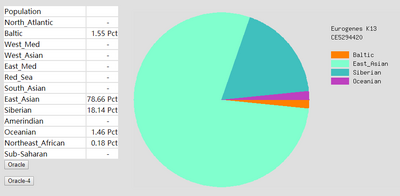
Figure 15: Ethnicity reports of sample DNA file 1 after degradation


-
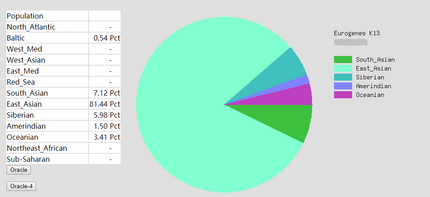
Figure 16: Ethnicity reports of sample DNA file 2 -
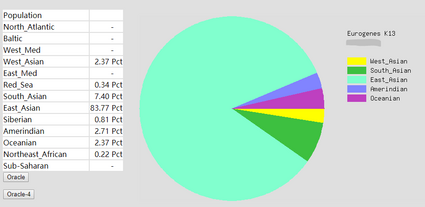
Figure 17: Ethnicity reports of sample DNA file 2 after degradation


To provide more evidences to prove this theory, several degradation algorithms introduced in section 5.2 have been applied and changes of ethnicity proportions during different degradation processes have been observed and recorded. 2 sample DNA reference files are degraded into 9 files at different levels from 90% to 10% SNPs remaining. The proportion of first 2 largest ethnicity regions of each degraded files have been plotted on line graphs. Figure 18 is the line graph that show how the means of ethnicity proportions change via the degradation process with standard error provided. As the graph shown, each region proportion fluctuate at a certain level. For instance the percentage of first region of sample 1 fluctuate at around 83% which is a close value to the original proportion 81.44%. However, error bars or standard errors of each region become larger, as more SNPs are removed, which indicate that as more SNPs being removed, the proportions presented in ethnicity reports become less accuracy. But in another case, the highest standard error for first and second region proportions of sample 1 and 2 are 1.32%, 1.41%, 1.33% and 1.03%. None of these standard errors exceed 1.5% which can be seen as an acceptable errors. Therefore the project conclude that when a large amount of SNPs are removed from a set of DNA data, the ethnicity report generated from the DNA data would be influenced, but the results are still acceptable for identifying owner’s ethnicity.

Conclusion
According to the observation of ethnicity change during the degradation process, as more amount of SNPs are removed from a complete human DNA reference file, the result of ethnicity report would be less accuracy. But for the largest and second largest ethnicity regions in the report are still reliable. Therefore the major ethnicity of Somerton Man is North Atlantic and Baltic.
Genetic diseases search
Aims
During this task, the team would focus on searching clinical effects of each available SNP and identify any possible genetic disease or physical characteristics that Somerton Man could have.
Methods
To search the clinical effects of SNPs, the team would develop a data mining program that collecting information in SNP database. Python language would be used for development since it is convenient for web development. The SNP database the project selected to use is dbSNP which is the largest database for nucleotide variations in the world, and is managed by the National Center for Biotechnology Information (NCBI) [11]. Figure 5.1 shows the information provided by dbSNP. The team would collect the clinical significance refers to each rsid in Somerton Man's file.

The program should be able to extract each non-empty SNP in Somerton Man's DNA reference file. With the API provided by dbSNP, connection to dbSNP should be established and each rsid of the extracted SNP should be sent. If the connection is successfully set up, dbSNP will send back the information of corresponding SNP in JSON format. Then the data sent back will be read and clinical information such as genetic disease name associated with the SNP will be recorded.
Result and discussion
With the support of data mining program, 613905 SNPs were searched in the database and 574 diseases were found. Figure 19 shows part of the genetic diseases outputs. As the figure shown, the program would record the rsid of SNP that the disease belonged to in rsid column. And column alleles present the change of nucleotide change of the SNPs. Moreover, dbSNP only provide a brief description of the clinical effects. More details are linked to another database called ClinVar which is a freely accessible, public database that provide medical reports of the relationships among human variants and phenotypes [12]. Therefore ClinVar Accession column is introduced to collect the ID of the recorded disease. This ID linked to the Clinvar database and allow the user to find a detailed medical report about the disease. Last but not least, the diseases names are recorded in disease name column. It is necessary to indicate that there are multiple diseases named with 'not specified' or 'not provided' which would require the user to find a detailed description of the disease in Clinvar. Unfortunately, none of diseases in the results relates or corresponds to Somerton Man's known appearance.

Task 5: Investigation on DNA matches
Aims
The aims of this task is to investigate what results if the DNA match services provide on GEDmatch are conducted on high quality DNA kits, and how the degradation could effect the match results.
Methods
In task 2, the project has conducted DNA match examination on Somerton Man’s DNA kit with multiple methods, but there is no match results for the his DNA reference file. In this task, one-to-many tool will be used again on 2 sample DNA files the project ordered from 23andMe, and the match results shall be recorded. Then the DNA match tests would be conducted on the degraded files created in task 3. The top 30 match results for each degraded DNA kit would be recorded and compare with the results of their original kit. A false positives and false negatives test would be conducted to show the change of match results during the degradation process. In this case, false positives would be match kits that are in the degraded kit’s match results but not in original kit’s result. And false negatives would present kits that are matched with original kit but not with the degraded one. An example is presented for a clear understanding. There are 5 kits A, B, C, D and E matched with the original kit, and kits A, B, C, M and N are matched with a degraded kit. Then the false positives for this degraded kit are D and E, and the false negatives are kits M and N. A line graphs of the number of false positives and false negatives against the percentage of SNPs removed will be created to show how degradation process effect the match results.
Results and discussion
Both DNA samples were successfully found their matched DNA kits in the database. Sample 1 have 8182 match kits and there are 5968 DNA files are found related to the sample 2. Top 30 match kits of sample 1 are shown in figure 20. The column Kit, Name and Email indicate the kit number, name of the kit and email of kit's owner. Column Total cM shows the total centimorgan which is a measure of genetic linkage between the 2 DNA kits. Note that the top 30 match kits are the kits with largest total centimorgan. Last but not least, the Overlap column present how many SNPs were used in the comparison between 2 kits.

Next, the top 30 match kits for each degraded DNA reference files are recorded and the false negatives and false positives are calculated. Since all degraded files except the degraded files with 10% SNPs remaining have more than 30 match kits, the number of false negatives and false positives are same. The degraded files with 10% SNPs remaining have no match results. There are 4 degradation strategies introduced in task 3, therefore 4 sets of false negatives and false positives are provided for analysis. Figure 21 present line graph of the number of false negatives and false positives against degradation levels. The number of false negatives and false positives are the mean of 4 sets of data. Degradation level of 10% SNP remaining is not involved in the graph due to 0 match result. Similar graph which was done by last year's project were shown in figure 22. The DNA sample used in figure 22 is a completely different one from the samples used in figure 21. According to both graphs, the number of false positives and false negatives for different DNA samples are not same. But the trend are similar. As more SNPs are removed, the amount of false positives and false negatives increases until 50% SNPs are removed. When there is more than half amount of SNPs being removed, the number of false positives and false negatives reaches maximum of 30 which indicate that the match results of original kits and degraded kits are totally different at these levels. These results show that as more SNPs removed from the original DNA reference file, the match results would be more inaccuracy. And when there is only half amount of SNPs remaining in the DNA kit, the match results would be totally different and be unreliable. Moreover, when 10% of SNPs are removed, more than half of match results would be different which indicates that even a small amount of SNPs being removed could result a huge difference in DNA match test.


Conclusion
According to the findings in this task, the project can conclude that it would require a high level quality of DNA which would be at least more than 90% SNPs are available in the DNA reference file to receive a reliable DNA match results. Only a small amount of SNPs in the DNA file are changed could result a significant affect on DNA match results. In another case, if the Somerton Man's DNA reference file is available to be recovered to more than 20% SNPs remaining, there could be DNA kits found related to him. And If the Somerton Man's DNA kit could be recoverd to a level of 60% SNPs remaining, part of his DNA match results can be reliable.
Project Management
Budget
There are $250 budgets assigned to each member in the project, in which is $500 budgets in total for the project. Most budgets are spent on ordering 2 DNA kits from 23andme company for DNA testing. The details are shown in the table below. There is a plan on spending the rest of budgets on purchasing the advance services provided on GEDmatch. But the team is still evaluating demand of using these services.

Risk Management
The risk assessment table are listed below. Several risks occurred during the progress. One of the group member was absent in the meeting several times due to time clash. But there is always at least one member attend the meeting with the supervisor. Members sometimes misunderstand assigned task, but issues were always fixed in the meeting in the following week.

Conclusions
The Somerton Man's DNA reference file provided to the project contain 613905 SNPs, but only 2.08% of SNPs are not empty and be able for DNA analysis. With such low proportion of available SNPs, limited DNA analysis techniques can be conducted on the file. Unfortunately, there is no DNA kits that matched with Somerton Man's DNA kit were found in GEDmatch database. According to the result of task 2 and task 4, the degradation process would have huge effect on the match results of a human DNA data. And it is impossible to recover Somerton Man's DNA by implementing simple recovery methods such as replacing empty SNPs with random base pairs or homozygous pairs. But if some reliable recovery strategies were introduced which have not been determined yet and allow Somerton Man's DNA to be recovered to more than 60% SNPs, then his relatives may be discovered. Moreover, it shows that Somerton Man comes from Europe. To be specific, his ethnicity is about 36.21% North Atlantic and 20.44% Baltic. As for the genetic disease, 574 diseases were found, but there is no disease was found that related to his known appearance. So far, that is what the project can found with Somerton Man's DNA data. There is no clear clue that can lead to his identity. Who the Somerton Man is will still be an unsolved mystery.
Future Work
So far most work that can operated with the Somerton Man's DNA. Several points can be introduced to improve the outcome of the project. Firstly, there are only 2 DNA samples in this project for analysis. If more sample DNA reference files can be collected, then the analysis on degradation can be more reliable. Also, the ethnicity of 2 DNA samples are Asian, but the Somerton Man's ethnicity has higher chance to be European. Therefore, if the project can have several European DNA data, more reliable DNA analysis can be done. Moreover, another future job can be done by this project is to recover the Somerton Man's DNA data by inserting SNPs that are common among European. This could require a large amount of DNA samples for discovering common SNPs which could be a challenge. Last but not least, finding a higher quality DNA data of Somerton Man would be the best way to identify the man, since higher quality DNA can allow more genetic examinations to be conducted.
Reference
[1]Bineth, J, "Somerton Man: One of Australia's most baffling cold cases could be a step closer to being solved" This Is About, 13 December 2017. [online] Available at: https://www.abc.net.au/news/2017-12-14/somerton-man-cold-case-could-be- one-step-closer-to-solved/9245512 [Accessed 1 Jun. 2019].
[2]U.S. National Library of Medicine, "What is DNA?",U.S. National Library of Medicine, May. 28, 2019. [online] Available at: https://ghr.nlm.nih.gov/primer/basics/dna [Accessed 2 Jun. 2019].
[3]U.S. National Library of Medicine, "What is a chromosome?",U.S. National Library of Medicine, May. 28, 2019. [online] Available at: https://ghr.nlm.nih.gov/primer/basics/chromosome [Accessed 2 Jun. 2019].
[4]U.S. National Library of Medicine, "How many chromosomes do people have?",U.S. National Library of Medicine, May. 28, 2019. [Online]. Available: https://ghr.nlm.nih.gov/primer/basics/howmanychromosomes. [Accessed: 02- Jun- 2019].
[5]U.S. National Library of Medicine, "What are single nucleotide polymorphisms (SNPs)?",U.S. National Library of Medicine, May. 28, 2019. [Online]. Available: https://ghr.nlm.nih.gov/primer/genomicresearch/snp. [Accessed: 02- Jun- 2019].
[6]G. Shaw. “Polymorphism and Single nucleotide polymorphisms (SNPs)” Science Made Simple, Vol. 112, pp.664-665 2013.
[7]“DEAD MAN FOUND LYING ON SOMERTON BEACH” The News, December 1, 1948, p. 1 [online]. Available: https://trove.nla.gov.au/newspaper/article/129897161. [Accessed: 03- Jun- 2019].
[8]“Cryptic Note On Body” The News, June 6, 1949, p. 1 [online]. Available: https://trove.nla.gov.au/newspaper/article/36371152. [Accessed: 03- Jun- 2019].
[9]"Raw Data Technical Details", 23andMe, 2019. [Online]. Available: https://customercare.23andme.com/hc/en-us/articles/115004459928-Raw-Data- Technical-Details. [Accessed: 04- Jun- 2019].
[10][10]S. Zhang, "The Coming Wave of Murders Solved by Genealogy", The Atlantic, 2019. [Online]. Available: https://www.theatlantic.com/science/archive/2018/05/the-coming-wave-of- murders-solved-by-genealogy/560750/. [Accessed: 04- Jun- 2019].
[11]"General Information about dbSNP as a Database Resource", Center for Biotechnology Information (US), 2005. [Online]. Available: https://www.ncbi.nlm.nih.gov/books/NBK44469/. [Accessed: 06- Jun- 2019].
[12]Landrum, M., Lee, J., Riley, G., Jang, W., Rubinstein, W., Church, D. and Maglott, D. (2013). ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Research, 42(D1), pp.D980-D985.
[13]Chick, H. (2017). Finally! A Gedmatch Admixture Guide!. [Blog] genealogical musings. Available at: https://genealogical-musings.blogspot.com/2017/04/finally-gedmatch-admixture-guide.html [Accessed 29- Oct- 2019].
[14]Chen, J. and Seroka, A. (2018). Cipher cracking Final Report/Thesis 2018. [online] Eleceng.adelaide.edu.au. Available at: https://www.eleceng.adelaide.edu.au/personal/dabbott/wiki/index.php/Final_Report/Thesis_2018 [Accessed 1 Nov. 2019].
[15]Davidski. (2019). K13_population_averages. [online] Available at: https://docs.google.com/spreadsheets/d/1Oz6P5-SVEJciPX1TciGe-zoqA5JtOGIMG7nh-rCOj0c/edit#gid=804264822 [Accessed 1 Nov. 2019].
[16]Inside Story, presented by Stuart Littlemore, ABC TV, screened at 8 pm, Thursday, 24th August, 1978